






�r�g��2024-05-19 ��Դ���ϷʾWhfw.cc ���ߣ�hfw.cc ��Ҫ�m�e

|

|

|

|

|

|

|
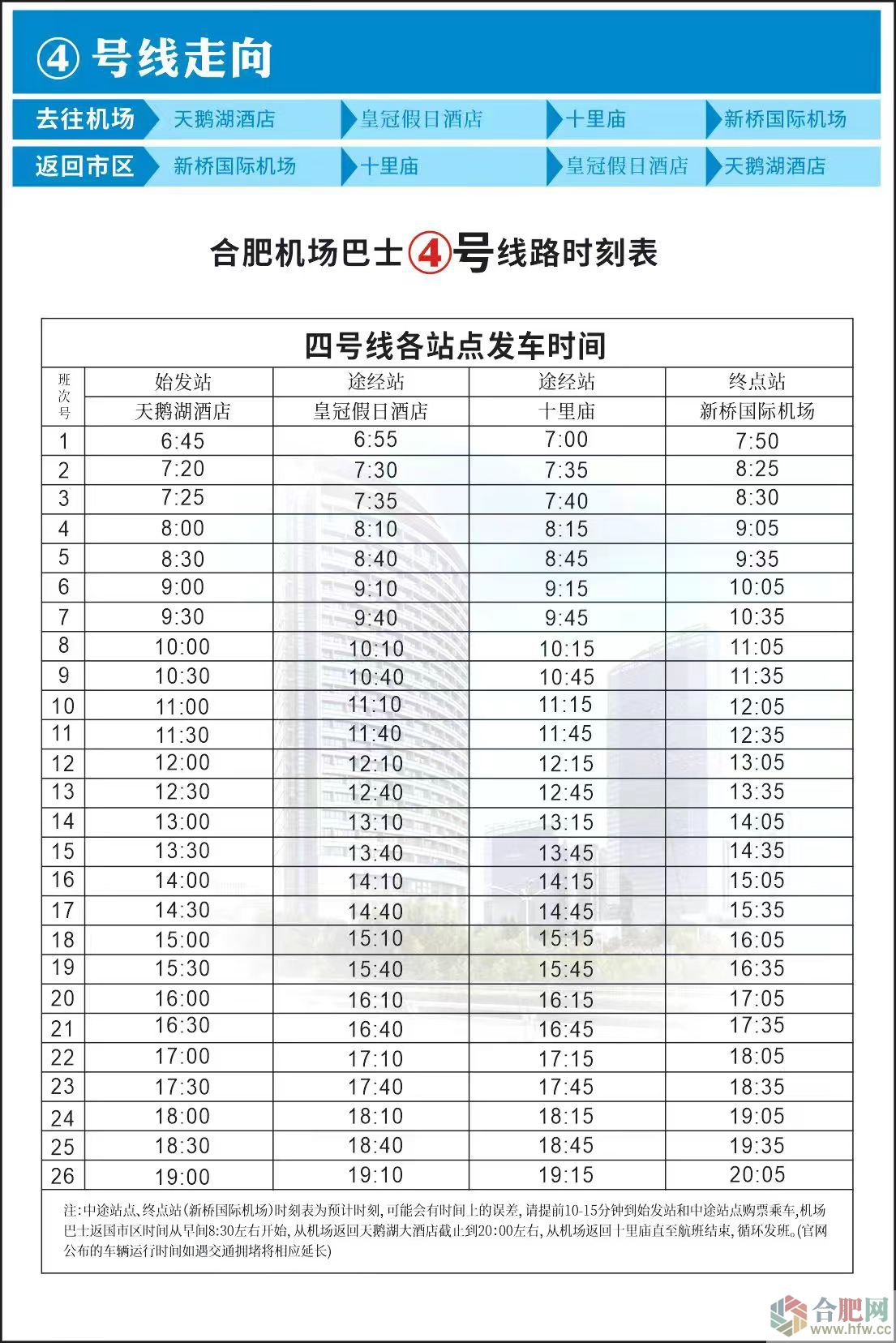
|
 |
������C�a �����W퓰���� Ŀ䛾W ���оW
Copyright © 2025 hfw.cc Inc. All Rights Reserved. �ϷʾW �������
ICP��06013414̖-3 ������ 42010502001045

